How To Use How To Interpret
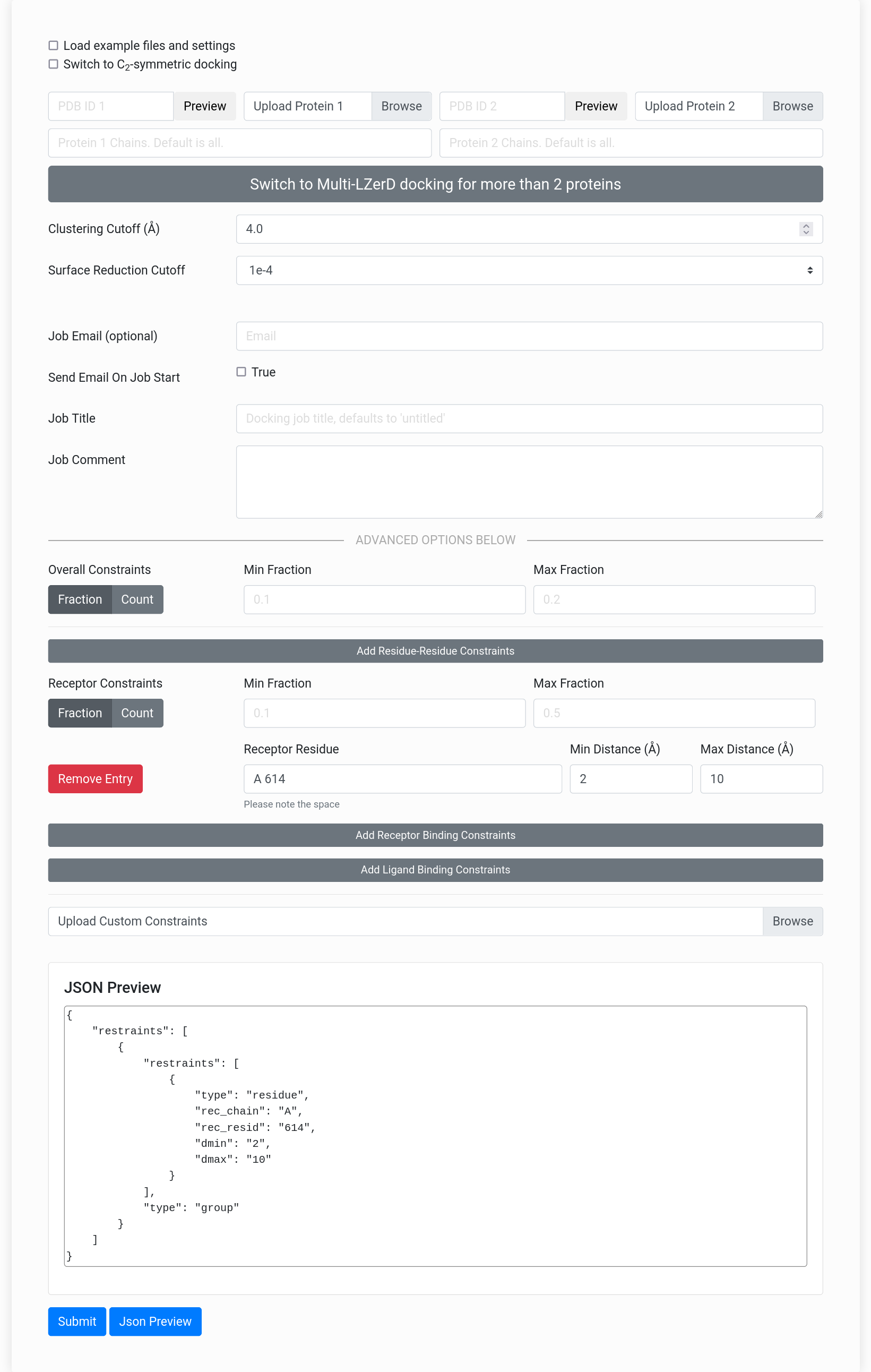 Figure On Right: The LZerD webserver input form.
Figure On Right: The LZerD webserver input form.
Registration
Click the button at the top right of the page to register for an account. Registered users will have the following benefits:- A list of submitted jobs will be easily viewable under the "Find a Job" tab
- Submitted jobs will remain for at least three months
- They will have to keep track of job links themselves
- Submitted jobs will be deleted after two weeks
Basic Usage
The LZerD webserver can accept fine-grained residue interaction constraints, but these are not necessary to perform docking. You can use LZerD simply by uploading your subunit structures in the PDB format through the "Upload Protein 1" and "Upload Protein 2" fields on the input page. Alternatively, you can specify a valid PDB ID to automatically fetch that structure. If you wish to only include certain chain IDs from your structure in the docking, you can specify them in the "Chains" fields.After uploading the two subunit structures, you can skip the rest of the form and click "Submit". It is recommended (but not required) that you fill in your email address to receive a notification of job completion. Job completion time depends on the number of queued jobs and the subunit size, but jobs typically complete within 8 hours after they begin running. Jobs may take up to a day to begin running, depending on the number of jobs in the queue. Unfinished jobs will display a status indicating the progress of the job. This status updates on docking start, clustering start, scoring start, and job completion. The status messages are enumerated at the bottom of the page. The Clustering Cutoff and Surface Reduction Cutoff can be used to reduce the computational time if necessary. The Clustering Cutoff controls the allowed root-mean-square deviation similarity between output models. Typically, the default of 4 Å reduces the number of models to a few thousand to a few tens of thousands, depending on the proteins; this number can be reduced by raising the cutoff. Surface Reduction Cutoff controls the number of docking sample points on each molecular surface by setting how close sample points can be before they are merged; setting this to a smaller value will result in a coarser docking search typically yielding a smaller number of models more quickly.
Advanced Usage
Following information from the literature or from experiments you may have performed, you may wish to only receive models with certain residue-residue interaction (or non-interaction) features. The LZerD webserver supports specifying fine grained residue interaction constraints. The four supported constraint types are described below.Overall residue constraints
Residue-residue distance
A residue-residue distance constraint restricts output to models where the distance between the specified residues falls within the specified minimum and maximum in angstroms. Leaving the minimum distance blank defaults to no minimum, while leaving the maximum distance blank defaults to no maximum. Residue-residue constraints can be used to specify interacting as well as non-interaction, depending on the minimum and maximum distances given. For example, to specify that two residues must interact, you can leave the minimum blank and set the maximum to e.g. 5 Å. To instead specify non-interaction, you can leave the maximum blank and instead set the minimum to e.g. 5 Å. Residues can be specified by their chain ID and residue number (e.g. B 115), which can include a residue insertion code (e.g. B 115A). For purposes of residue constraint satisfaction, all alternate atom locations (i.e. "altlocs") except the first are ignored.Receptor protein site
Protein 1 uploaded is considered as the receptor protein. A receptor site constraint is like a residue-residue constraint, except only a receptor residue is specified, and the distance cutoffs apply to the nearest ligand residue in a given model.Ligand protein site
Protein 2 uploaded is considered as the ligand protein. A ligand site constraint is exactly like a receptor site constraint, except a ligand residue is specified.Groups
A group constraint bundles together any number of other constraints (including other group constraints). Groups allow you to specify multiple constraints and specify that a certain number or fraction of them should be satisfied. This can be used to create soft restraints (e.g. set the cutoff to 50% of child constraints) and boolean constraints (e.g. a cutoff of 100% requires all child constraints to be satisfied, while a cutoff of 1 merely requires that any child constraint be satisfied).Constraint input using JSON
Instead of using the graphical interface, you can supply constraints to the LZerD server by uploading appropriately formatted JSON file. You can see the equivalent JSON encoding of constraints specified in the GUI by clicking the "JSON Preview" button. Below is an example of JSON file contents (downloadable here) which demonstrates the format. There are two types of constraint objects, residue constraints and group constraints. A residue constraint specifies either a receptor or ligand residue or both, as well as a distance range. This type of constraint is satisfied if distance between the closest pair of heavy atoms between the two residues falls within the distance range. If either the receptor or the ligand is not specified, the entire subunit is considered instead of a single residue. This type of constraint is the basic unit for individual residue-residue or binding site constraints specified by the graphical interface. Group constraints specify a list of constraints (both residue constraints and other group constraints are allowed) along with a minimum and maximum amount of listed constraints to satisfy. These amounts may be specified either by actual number of constraints or by the fraction of constraints, just like in the graphical interface.In this example, four residue-level constraints are specified and combined into groups. Either or both of the residue pairs [receptor chain X residue 1 and ligand chain Y residue 2] or [receptor chain Q residue 1 and ligand chain W residue 2] should be in contact; this is achieved by setting the minimum fraction of satisfied constraints to be greater than zero via
"min_fraction": "0.3", meaning any nonzero number of satisfied sub-constraints satisfies the group.
"max_fraction": "1.0"simply means that all child constraints are allowed to be satisfied.
"max_number": "99", "min_number": "1"accomplishes the same task here, except using exact count cutoffs instead of fractions. Additionally, receptor chain A residue 1 and ligand chain A residue 2 should each be contacting another subunit. The condition of residue "contact" is set using the the
dminand
dmaxfields, which set the range of allowed residue-residue distances; thus,
"dmin": "0", "dmax": "5"sets the allowed distance range to 0 Å to 5 Å. The
"COMMENT"lines below highlight the details of the format.
{
"COMMENT": "This group specifies the number of component constraints which should be satisfied.",
"type": "group",
"COMMENT": "This list contains the component constraints of the group.",
"restraints": [
{
"restraints": [
{
"COMMENT": "This residue constraint specifies a specific pair of residues which should be within a certain distance range.",
"type": "residue",
"rec_chain": "X",
"rec_resid": "1",
"lig_chain": "Y",
"lig_resid": "2",
"dmin": "0",
"dmax": "5"
},
{
"type": "residue",
"rec_chain": "Q",
"rec_resid": "1",
"lig_chain": "W",
"lig_resid": "2",
"dmin": "0",
"dmax": "5"
}
],
"type": "group",
"COMMENT": "This group specifies the fraction of component constraints which should be satisfied.",
"max_fraction": "1.0",
"min_fraction": "0.3"
},
{
"restraints": [
{
"COMMENT": "This residue constrain specifies only a receptor residue, and will be satisfied if the distance to the closest ligand residue falls within the specified range.",
"type": "residue",
"rec_chain": "A",
"rec_resid": "1",
"dmin": "0",
"dmax": "5"
}
],
"max_number": "99",
"min_number": "1",
"type": "group"
},
{
"restraints": [
{
"type": "residue",
"lig_chain": "A",
"lig_resid": "2",
"dmin": "0 ",
"dmax": "5"
}
],
"type": "group",
"max_number": "99",
"min_number": "1"
}
]
}
Job Status Details
Jobs which have not yet finished may have the followed status messages attached:- Job is queued.
- Job is running.
- Job is running on remote cluster.
- Job is running (preprocessing).
- Job is running (docking search).
- Job is running (clustering).
- Job is running (scoring).
- Job is running (pairwise docking search).
- Job is running (multiple docking search).
Video Tutorial
This video tutorial describes the docking method and demonstrates how to use the server: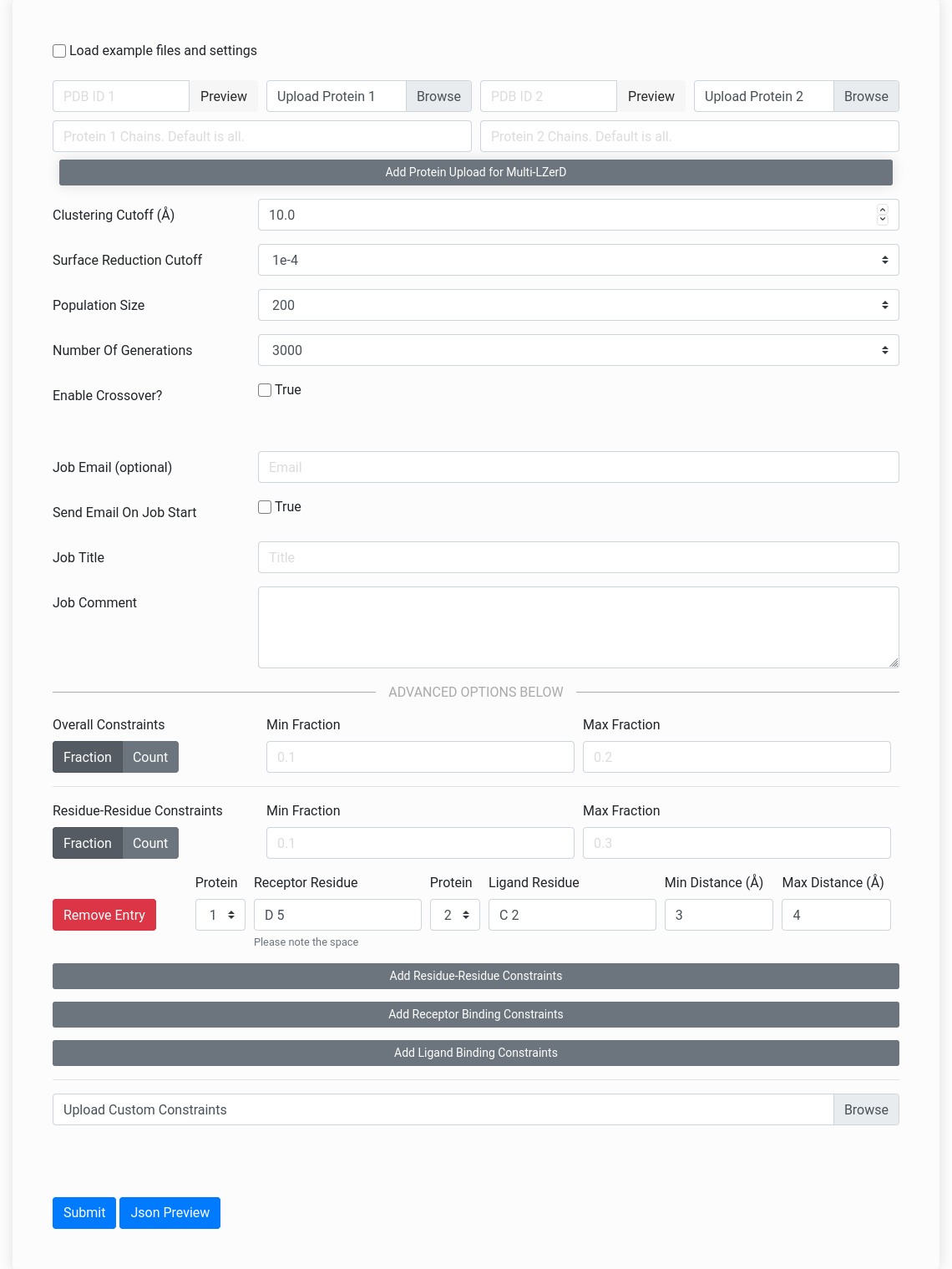 Figure On Right: The Multi-LZerD webserver input form.
Figure On Right: The Multi-LZerD webserver input form.
Registration
Click the button at the top right of the page to register for an account. Registered users will have the following benefits:- A list of submitted jobs will be easily viewable under the "Find a Job" tab
- Submitted jobs will remain for at least three months
- They will have to keep track of job links themselves
- Submitted jobs will be deleted after two weeks
Basic Usage
The Multi-LZerD webserver can accept fine-grained residue interaction constraints, but these are not necessary to perform docking. You can use Multi-LZerD simply by uploading your subunit structures in the PDB format through the "Upload Protein 1", "Upload Protein 2", etc. fields on the input page. Alternatively, you can specify a valid PDB ID to automatically fetch that structure. If you wish to only include certain chain IDs from your structure in the docking, you can specify them in the "Chains" fields. You can increase and decrease the number of input structures by clicking the "Add Protein" and "Remove Protein" buttons respectively. The webserver supports docking up to six subunits simultaneously. A standalone executable version which supports docking more than six subunits is available for download here.After uploading the two subunit structures, you can skip the rest of the form and click "Submit". It is recommended (but not required) that you fill in your email address to receive a notification of job completion. Job completion time depends on the number of queued jobs, the subunit size, and the number of input structures, but jobs typically complete within 48 hours after they begin running. Jobs may take up to a few days to begin running, depending on the number of pending jobs in the queue. Unfinished jobs will display a status indicating the progress of the job. This status updates on pairwise docking start, multiple docking start, scoring start, and job completion. The status messages are enumerated at the bottom of the page.
Advanced Usage
Multi-LZerD allows users to control the number of output models through the "Population Size" parameter. The "Number of Generations" parameter controls how extensively the docking search space is explored. The "Clustering Cutoff" controls the level of diversity in the output model set. Following information from the literature or from experiments you may have performed, you may wish to only receive models with certain residue-residue interaction (or non-interaction) features. The Multi-LZerD webserver supports specifying fine grained residue interaction constraints. The three supported constraint types are described below.Overall residue constraints
Residue-residue distance
A residue-residue distance constraint restricts output to models where the distance between the specified residues falls within the specified minimum and maximum in angstroms. Leaving the minimum distance blank defaults to no minimum, while leaving the maximum distance blank defaults to no maximum. Residue-residue constraints can be used to specifiy interacting as well as non-interaction, depending on the minimum and maximum distances given. For example, to specify that two residues must interact, you can leave the minimum blank and set the maximum to e.g. 5 Å. To instead specify non-interaction, you can leave the maximum blank and instead set the minimum to e.g. 5 Å. Residues can be specified by their input structure number, chain ID, and residue number (e.g. B 115), which can include a residue insertion code (e.g. B 115A). For purposes of residue constraint satisfaction, all alternate atom locations (i.e. "altlocs") except the first are ignored.Receptor protein site
For each receptor site constraint, the specified structure is considered as the receptor protein. A receptor site constraint is like a residue-residue constraint, except only a receptor residue is specified, and the distance cutoffs apply to the nearest residue belonging to a different input structure in a given model.Groups
A group constraint bundles together any number of other constraints (including other group constraints). Groups allow you to specify multiple constraints and specify that a certain number or fraction of them should be satisfied. This can be used to create soft restraints (e.g. set the cutoff to 50% of child constraints) and boolean constraints (e.g. a cutoff of 100% requires all child constraints to be satisfied, while a cutoff of 1 merely requires that any child constraint be satisfied).Constraint input using JSON
Instead of using the graphical interface, you can supply constraints to the LZerD server by uploading appropriately formatted JSON file. You can see the equivalent JSON encoding of constraints specified in the GUI by clicking the "JSON Preview" button. Below is an example of JSON file contents (downloadable here) which demonstrates the format. There are two types of constraint objects, residue constraints and group constraints. A residue constraint specifies either a receptor or ligand residue or both, as well as a distance range. This type of constraint is satisfied if distance between the closest pair of heavy atoms between the two residues falls within the distance range. If either the receptor or the ligand is not specified, the entire subunit is considered instead of a single residue. This type of constraint is the basic unit for individual residue-residue or binding site constraints specified by the graphical interface. Group constraints specify a list of constraints (both residue constraints and other group constraints are allowed) along with a minimum and maximum amount of listed constraints to satisfy. These amounts may be specified either by actual number of constraints or by the fraction of constraints, just like in the graphical interface.In this example, four residue-level constraints are specified and combined into groups. Either or both of the residue pairs [subunit 1 chain X residue 1 and subunit 2 chain Y residue 2] or [subunit 1 chain Q residue 1 and subunit 2 chain W residue 2] should be in contact; this is achieved by setting the minimum number of satisfied constraints to be 1 via the min_number field, meaning any nonzero number of satisfied sub-constraints satisfies the group. min_number being set to 1 simply means that all child constraints are allowed to be satisfied. Additionally, subunit 1 chain A residue 1 and subunit 2 chain A residue 2 should each be contacting another subunit. The condition of residue "contact" is set using the the
dminand
dmaxfields, which set the range of allowed residue-residue distances; thus,
"dmin": "0", "dmax": "5"sets the allowed distance range to 0 Å to 5 Å. The
"COMMENT"lines below highlight the details of the format.
{
"COMMENT": "This group specifies the number of component constraints which should be satisfied.",
"type": "group",
"COMMENT": "This list contains the component constraints of the group.",
"restraints": [
{
"restraints": [
{
"COMMENT": "This residue constraint specifies a specific pair of residues which should be within a certain distance range.",
"type": "residue",
"rec_subunit_id": "1"
"rec_chain": "X",
"rec_resid": "1",
"lig_subunit_id": "2"
"lig_chain": "Y",
"lig_resid": "2",
"dmin": "0",
"dmax": "5"
},
{
"type": "residue",
"rec_subunit_id": "1"
"rec_chain": "Q",
"rec_resid": "1",
"lig_subunit_id": "2"
"lig_chain": "W",
"lig_resid": "2",
"dmin": "0",
"dmax": "5"
}
],
"type": "group",
"COMMENT": "This group specifies the fraction of component constraints which should be satisfied.",
"max_number": "99",
"min_number": "1"
},
{
"restraints": [
{
"COMMENT": "This residue constraint specifies only a receptor residue, and will be satisfied if the distance to the closest ligand residue falls within the specified range.",
"type": "residue",
"rec_subunit_id": "1"
"rec_chain": "A",
"rec_resid": "1",
"dmin": "0",
"dmax": "5"
}
],
"max_number": "99",
"min_number": "1",
"type": "group"
},
{
"restraints": [
{
"type": "residue",
"lig_subunit_id": "2"
"lig_chain": "A",
"lig_resid": "2",
"dmin": "0 ",
"dmax": "5"
}
],
"type": "group",
"max_number": "99",
"min_number": "1"
}
]
}
Video Tutorial
This video tutorial describes the docking method and demonstrates how to use the server:Job Status Details
Jobs which have not yet finished may have the followed status messages attached:- Job is queued.
- Job is running.
- Job is running on remote cluster.
- Job is running (preprocessing).
- Job is running (docking search).
- Job is running (clustering).
- Job is running (scoring).
- Job is running (pairwise docking search).
- Job is running (multiple docking search).
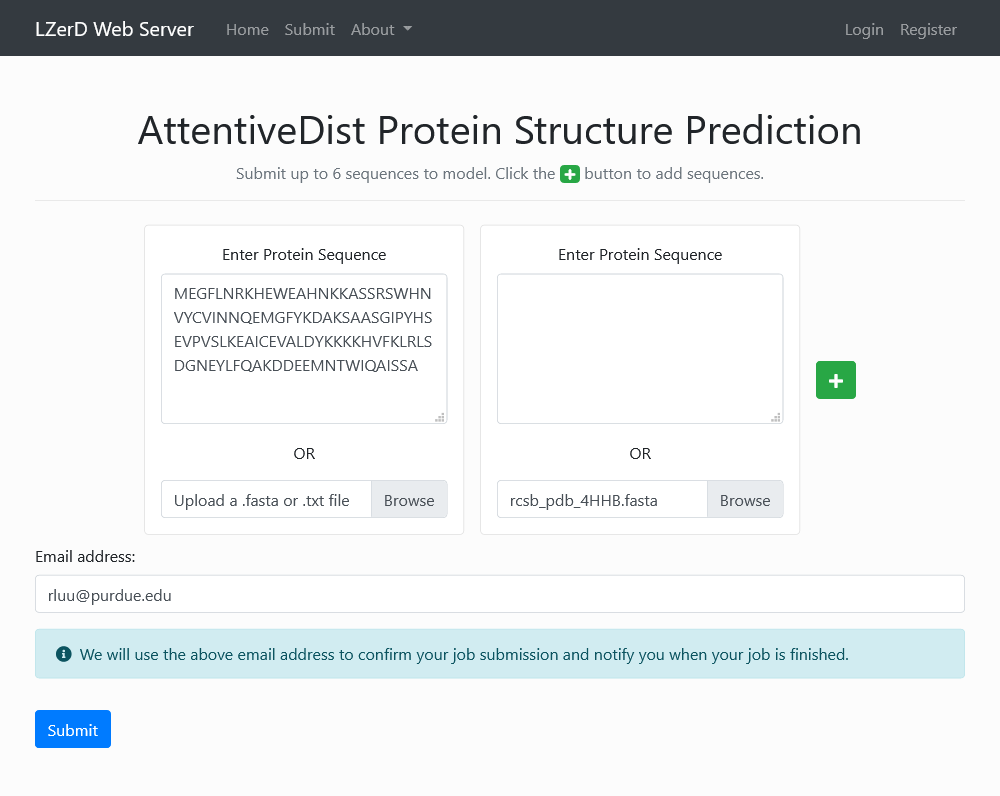
Submitting Sequences
Navigate to Home > Upload Protein Sequences or click here to begin submitting sequences to model using AttentiveDist. A sequence in FASTA format can be pasted into the text area or uploaded as a .fasta or .txt file. To model more than one sequence (up to six), click the button. A valid email address can be provided to confirm your job submission and notify you when your job is finished.
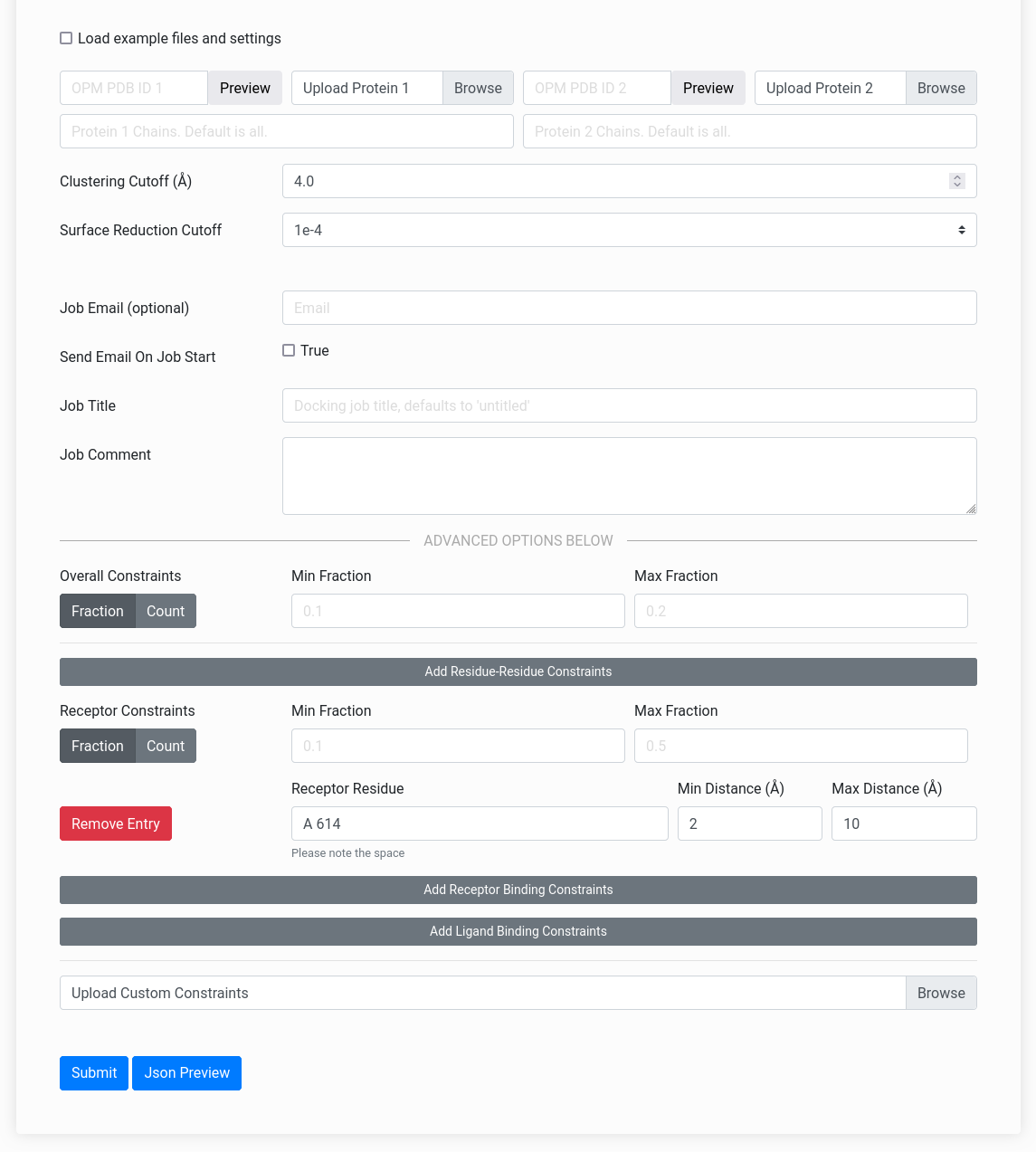 Figure On Right: The Mem-LZerD webserver input form.
Figure On Right: The Mem-LZerD webserver input form.
Registration
Click the button at the top right of the page to register for an account. Registered users will have the following benefits:- A list of submitted jobs will be easily viewable under the "Find a Job" tab
- Submitted jobs will remain for at least three months
- They will have to keep track of job links themselves
- Submitted jobs will be deleted after two weeks
Basic Usage
The Mem-LZerD webserver can accept fine-grained residue interaction constraints, but these are not necessary to perform docking. You can use Mem-LZerD simply by uploading your membrane-oriented subunit structures in the PDB format through the "Upload Protein 1" and "Upload Protein 2" fields on the input page. Alternatively, you can specify a valid PDB ID to automatically use the corresponding OPM database entry for that structure. If you wish to only include certain chain IDs from your structure in the docking, you can specify them in the "Chains" fields.After uploading the two subunit structures, you can skip the rest of the form and click "Submit". It is recommended (but not required) that you fill in your email address to receive a notification of job completion. Job completion time depends on the number of queued jobs and the subunit size, but jobs typically complete within 8 hours after they begin running. Jobs may take up to a day to begin running, depending on the number of jobs in the queue. Unfinished jobs will display a status indicating the progress of the job. This status updates on docking start, clustering start, scoring start, and job completion. The status messages are enumerated at the bottom of the page. The Clustering Cutoff and Surface Reduction Cutoff can be used to reduce the computational time if necessary. The Clustering Cutoff controls the allowed root-mean-square deviation similarity between output models. Typically, the default of 4 Å reduces the number of models to a few thousand to a few tens of thousands, depending on the proteins; this number can be reduced by raising the cutoff. Surface Reduction Cutoff controls the number of docking sample points on each molecular surface by setting how close sample points can be before they are merged; setting this to a smaller value will result in a coarser docking search typically yielding a smaller number of models more quickly.
Advanced Usage
Following information from the literature or from experiments you may have performed, you may wish to only receive models with certain residue-residue interaction (or non-interaction) features. The Mem-LZerD webserver supports specifying fine grained residue interaction constraints. The four supported constraint types are described below. Please note that if the constraints you specify are geometrically incompatible with the membrane orientation you provide, the output model set will be empty.Overall residue constraints
Residue-residue distance
A residue-residue distance constraint restricts output to models where the distance between the specified residues falls within the specified minimum and maximum in angstroms. Leaving the minimum distance blank defaults to no minimum, while leaving the maximum distance blank defaults to no maximum. Residue-residue constraints can be used to specify interacting as well as non-interaction, depending on the minimum and maximum distances given. For example, to specify that two residues must interact, you can leave the minimum blank and set the maximum to e.g. 5 Å. To instead specify non-interaction, you can leave the maximum blank and instead set the minimum to e.g. 5 Å. Residues can be specified by their chain ID and residue number (e.g. B 115), which can include a residue insertion code (e.g. B 115A). For purposes of residue constraint satisfaction, all alternate atom locations (i.e. "altlocs") except the first are ignored.Receptor protein site
Protein 1 uploaded is considered as the receptor protein. A receptor site constraint is like a residue-residue constraint, except only a receptor residue is specified, and the distance cutoffs apply to the nearest ligand residue in a given model.Ligand protein site
Protein 2 uploaded is considered as the ligand protein. A ligand site constraint is exactly like a receptor site constraint, except a ligand residue is specified.Groups
A group constraint bundles together any number of other constraints (including other group constraints). Groups allow you to specify multiple constraints and specify that a certain number or fraction of them should be satisfied. This can be used to create soft restraints (e.g. set the cutoff to 50% of child constraints) and boolean constraints (e.g. a cutoff of 100% requires all child constraints to be satisfied, while a cutoff of 1 merely requires that any child constraint be satisfied).Constraint input using JSON
Instead of using the graphical interface, you can supply constraints to the LZerD server by uploading appropriately formatted JSON file. You can see the equivalent JSON encoding of constraints specified in the GUI by clicking the "JSON Preview" button. Below is an example of JSON file contents (downloadable here) which demonstrates the format. There are two types of constraint objects, residue constraints and group constraints. A residue constraint specifies either a receptor or ligand residue or both, as well as a distance range. This type of constraint is satisfied if distance between the closest pair of heavy atoms between the two residues falls within the distance range. If either the receptor or the ligand is not specified, the entire subunit is considered instead of a single residue. This type of constraint is the basic unit for individual residue-residue or binding site constraints specified by the graphical interface. Group constraints specify a list of constraints (both residue constraints and other group constraints are allowed) along with a minimum and maximum amount of listed constraints to satisfy. These amounts may be specified either by actual number of constraints or by the fraction of constraints, just like in the graphical interface.In this example, four residue-level constraints are specified and combined into groups. Either or both of the residue pairs [receptor chain X residue 1 and ligand chain Y residue 2] or [receptor chain Q residue 1 and ligand chain W residue 2] should be in contact; this is achieved by setting the minimum fraction of satisfied constraints to be greater than zero via
"min_fraction": "0.3", meaning any nonzero number of satisfied sub-constraints satisfies the group.
"max_fraction": "1.0"simply means that all child constraints are allowed to be satisfied.
"max_number": "99", "min_number": "1"accomplishes the same task here, except using exact count cutoffs instead of fractions. Additionally, receptor chain A residue 1 and ligand chain A residue 2 should each be contacting another subunit. The condition of residue "contact" is set using the the
dminand
dmaxfields, which set the range of allowed residue-residue distances; thus,
"dmin": "0", "dmax": "5"sets the allowed distance range to 0 Å to 5 Å. The
"COMMENT"lines below highlight the details of the format.
{
"COMMENT": "This group specifies the number of component constraints which should be satisfied.",
"type": "group",
"COMMENT": "This list contains the component constraints of the group.",
"restraints": [
{
"restraints": [
{
"COMMENT": "This residue constraint specifies a specific pair of residues which should be within a certain distance range.",
"type": "residue",
"rec_chain": "X",
"rec_resid": "1",
"lig_chain": "Y",
"lig_resid": "2",
"dmin": "0",
"dmax": "5"
},
{
"type": "residue",
"rec_chain": "Q",
"rec_resid": "1",
"lig_chain": "W",
"lig_resid": "2",
"dmin": "0",
"dmax": "5"
}
],
"type": "group",
"COMMENT": "This group specifies the fraction of component constraints which should be satisfied.",
"max_fraction": "1.0",
"min_fraction": "0.3"
},
{
"restraints": [
{
"COMMENT": "This residue constrain specifies only a receptor residue, and will be satisfied if the distance to the closest ligand residue falls within the specified range.",
"type": "residue",
"rec_chain": "A",
"rec_resid": "1",
"dmin": "0",
"dmax": "5"
}
],
"max_number": "99",
"min_number": "1",
"type": "group"
},
{
"restraints": [
{
"type": "residue",
"lig_chain": "A",
"lig_resid": "2",
"dmin": "0 ",
"dmax": "5"
}
],
"type": "group",
"max_number": "99",
"min_number": "1"
}
]
}
Job Status Details
Jobs which have not yet finished may have the followed status messages attached:- Job is queued.
- Job is running.
- Job is running on remote cluster.
- Job is running (preprocessing).
- Job is running (docking search).
- Job is running (clustering).
- Job is running (scoring).
- Job is running (pairwise docking search).
- Job is running (multiple docking search).
Video Tutorial
This video tutorial describes the docking method and demonstrates how to use the server:Browser Compatibility
It is recommended that you use one of the following major web browsers to access the LZerD server:| OS | Version | Firefox | Chrome | Edge | Safari |
|---|---|---|---|---|---|
| Linux | CentOS 7 | ≥68 | ≥87 | n/a | n/a |
| Windows | ≥8.1 | ≥68 | ≥87 | ≥86 | n/a |
| MacOS | ≥Catalina | ≥68 | ≥87 | n/a | ≥13 |